
Epitheliale-mesenchymale overgang


De epitheliale-mesenchymale overgang (EMT) is een proces waarbij epitheelcellen hun celpolariteit en cel-celadhesie verliezen, en migrerende en invasieve eigenschappen krijgen om mesenchymatische stamcellen te worden; dit zijn multipotente stromacellen die kunnen differentiëren in verschillende celtypen. EMT is essentieel voor tal van ontwikkelingsprocessen, waaronder de vorming van mesoderm en de vorming van de neurale buis. Er is ook aangetoond dat EMT optreedt bij wondgenezing, bij orgaanfibrose en bij het initiëren van metastasen bij de progressie van kanker.
Deze overgang vindt plaats door het verlies van cadherine, zonula occludens, zonula adhaerens en desmosomen op de celmembranen van epitheelcellen. De oppervlaktemoleculen ondergaan endocytose en het cytoskelet van de microtubuli verliest vorm, waardoor het mesenchym langs de extracellulaire matrix (ECM) kan migreren. De epitheliale-mesenchymale overgang vindt plaats in embryonale cellen en vereist migratie door of over weefsel, en kan worden opgevolgd door een epitheliale-mesenchymale overgang voor het vormen van secundaire epitheelweefsels. Embryologische mesenchymale cellen brengen proteïne S100-A4 (S100A4) tot expressie, ook bekend als het fibroblast-specifiek eiwit, wat indicatief is voor hun gedeelde eigenschappen met de migrerende volwassen fibroblasten, en c-Fos, een oncogen geassocieerd met de downregulering van het epithele cadherine.
De epitheliale-mesenchymale overgang werd voor het eerst herkend als een kenmerk van embryogenese door Betty Hay in de jaren tachtig.[1][2] EMT en het omgekeerde proces ervan, MET (mesenchymale-epitheliale overgang) zijn van cruciaal belang voor de ontwikkeling van veel weefsels en organen in het zich ontwikkelende embryo, en voor talrijke embryonale gebeurtenissen zoals gastrulatie, vorming van de neurale lijst, vorming van hartkleppen, ontwikkeling van het secundaire gehemelte en myogenese.[3] Mesenchymcellen daarentegen missen deze polarisatie, hebben een spoelvormige morfologie en interageren alleen met elkaar via centrale plaatsen.[4] Epitheelcellen brengen hoge niveaus van E-cadherine tot expressie, terwijl mesenchymcellen die van N-cadherine, fibronectine en vimentine tot expressie brengen. EMT brengt dus diepgaande morfologische en fenotypische veranderingen in een cel met zich mee.[5]
Epitheel- en mesenchymcellen
[bewerken | brontekst bewerken]Epitheelcellen kunnen niet migreren en worden gekenmerkt door een
- apico-basale polariteit met binding door een basaal membraan,
- zonula occludens,
- gap junctions,
- zonula adhaerens en
- expressie van celadhesiemerkers zoals E-cadherine
Mesenchymale cellen kunnen wel migreren en
- hebben geen volwassen celadhesie,
- kunnen binnendringen via de extracellulaire matrix en
- genetische merkers tot expressie brengen zoals vimentine, fibronectine, N-cadherine, Twist en Snail
De mesenchymale-epitheliale overgang speelt ook een cruciale rol bij veranderingen van de stofwisseling en epigenetische modificaties. Over het algemeen worden epitheel-geassocieerde genen upstream en worden mesenchym-geassocieerde genen downstream tijdens het MET-proces tot expressie gebracht.
EMT-typen
[bewerken | brontekst bewerken]
Op basis van de biologische context is EMT onderverdeeld in 3 typen:
- ontwikkelingsstoornissen (Type 1),
- fibrose[6] en wondgenezing (Type 2) en
- kanker (Type 3).[7][8][9]
Type 1
[bewerken | brontekst bewerken]Type 1 EMT wordt in verband gebracht met de innesteling en ontwikkeling van embryo's, evenals met de vorming van meerdere organen, en veroorzaakt geen fibrose en induceert ook geen invasief fenotype. Deze EMT is nodig voor de productie van mesenchymcellen (primair mesenchym) die in staat zijn om achtereenvolgens een MET te ondergaan om secundair epitheel te creëren. Embryonale ontwikkeling is een complexe gebeurtenis waarbij zowel EMT als MET nodig zijn voor de uiteindelijke differentiatie van gespecialiseerde celtypen en voor de vorming van de driedimensionale organisatie van de organen. Cellen van het primair mesenchym hebben verhoogde migratiekenmerken. Op biochemisch niveau wordt de EMT gecorreleerd met gastrulatie gecoördineerd door canonieke Wnt-signalering. EMT geassocieerd met gastrulatie wordt gereguleerd door transcriptiefactoren van Snail, eomesodermine (Eomes) en mesoderm posterior proteine (Mesp's) en ook door Wnt's, dat samenwerkt met fibroblastgroeifactor (FGF)-receptoren voor het reguleren van EMT gekoppeld aan gastrulatie. Slak onderdrukt E-cadherine en bevordert EMT gemedieerd door celadhesiemoleculen zoals occludinen en claudines en door polariteitsgenen, zoals Discs large (Dlg) en Crumbs homoloog 3 (Crb3). Tijdens de embryonale vorming geeft een EMT, inclusief de epitheelcellen van het neuro-ectoderm, aanleiding tot migrerende neurale lijstcellen. De EMT's die verband houden met wondgenezing, weefselregeneratie en orgaanfibrose worden geclassificeerd als type 2-EMT's.[10]
Type 2
[bewerken | brontekst bewerken]
Type 2 EMT-gebeurtenissen vinden plaats als onderdeel van een herstel-geassocieerd proces waarbij de epitheelcellen differentiëren tot nieuwe fibroblastachtige cellen om weefsels te herstellen na verwonding en ontstekingsschade. Deze cellen hebben een epitheelspecifieke morfologie en moleculaire merkers, zoals cytokeratine en E-cadherine, maar brengen tegelijkertijd het ferroptosis-suppressoreiwit 1 (FSP1, een S100-klasse van cytoskeleteiwit) mesenchymale merker en alfa-gladde spieractine (α- SMA) tot expressie. Type 2 EMT's worden in verband gebracht met ontstekingen en worden beëindigd zodra het herstel is voltooid en de ontsteking is verminderd.
Fibrose onderscheidt zich door een overmatige afzetting van vezelig bindweefsel in een orgaan. Gedefinieerd door de pathologische accumulatie van ECM-componenten, die na verloop van tijd leiden tot de ontwikkeling van littekenweefsel en uiteindelijk tot orgaandisfunctie en -falen. Type 2 EMT is gekoppeld aan weefselherstelreacties zoals fibrose. Om beschadigde weefsels te genezen, geeft type 2 EMT aanleiding tot myofibroblasten uit epitheel; de genezingsgebeurtenis wordt beschouwd als herstellende fibrose als het letsel matig en acuut is. In plaats daarvan veroorzaakt de abnormale vorming van myofibroblasten aanhoudende chronische ontstekingen een progressieve fibrose die leidt tot orgaanparenchymale vernietiging als gevolg van een overmatige ECM-afzetting. Weefselfibrose is dus een onophoudelijke vorm van wondgenezing die het gevolg is van een afwijkend ontstekingsproces. FSP1, α-SMA en collageen I heeft betrouwbare merkers opgeleverd om de mesenchymale producten te karakteriseren die worden gegenereerd door de EMT's die optreden tijdens de ontwikkeling van fibrose in verschillende organen. Deze merkers zijn, samen met discoidin-domeinreceptortyrosinekinase 2 (DDR2), vimentine en desmine, gebruikt om epitheelcellen van de nieren, lever, longen en darmen te identificeren die zich midden in een EMT bevinden die verband houdt met chronische ontstekingen. Verschillende in vitro en in vivo onderzoeken hebben gemeld dat EMT betrokken is bij de fibrogenese van cruciale organen, zoals de nier, lever, long en darm. Studies uitgevoerd in een transgeen muismodel hebben bijvoorbeeld de betrokkenheid van EMT bij nierfibrose bevestigd en aangetoond dat >30% van de nieuwe fibroblasten afkomstig is van lokale EMT.[10]
Type 3
[bewerken | brontekst bewerken]Type 3 EMT komt voor in neoplastische cellen die genetische en epigenetische modificaties hebben ondergaan, vooral in genen die de vorming van gelokaliseerde tumoren bevorderen. Verschillende onderzoeken hebben aangetoond dat carcinoomcellen een mesenchymaal fenotype kunnen verwerven en mesenchymale merkers zoals α-SMA, FSP1, vimentine en desmine tot expressie kunnen brengen. Dergelijke cellen blijven epitheelmerkers tot expressie brengen, maar er zijn al nieuwe mesenchymmerkers verkregen. Deze veranderingen, die oncogenen en tumorsuppressorgenen modificeren, werken samen met het EMT-regulerende circuit om andere effecten teweeg te brengen dan die gerapporteerd in type 1 en type 2 EMT's. De migrerende kankercellen geproduceerd door type 3 EMT bevorderen bijvoorbeeld secundaire tumoren in afgelegen weefsels met epitheliale fenotypes. Dit suggereert dat de omkeerbaarheid van EMT, die een cruciale factor is bij de embryogenese, ook een sleutelrol speelt bij de ontwikkeling van secundaire metastatische knobbeltjes. Daarom is type 3 EMT noodzakelijk om metastatische kankercellen in staat te stellen aan apoptose te ontsnappen en de expressie van oncogenen te induceren.[10]
Inductoren
[bewerken | brontekst bewerken]
Het verlies van E-cadherine wordt beschouwd als een fundamentele gebeurtenis bij EMT. Veel transcriptiefactoren (TF's) die E-cadherine direct of indirect kunnen onderdrukken, kunnen worden beschouwd als EMT-TF (EMT-inducerende TF's). SNAI1/Snail 1, SNAI2/Snail 2 (ook bekend als Slug), ZEB1, ZEB2, TCF3 en KLF8 (Kruppel-achtige factor 8) kunnen binden aan de E-cadherine-promotor en de transcriptie ervan onderdrukken, terwijl factoren zoals Twist, Goosecoid TCF4 (ook bekend als E2.2), homeobox-eiwit SIX1 en FOXC2 (vork-head box-eiwit C2) onderdrukken indirect E-cadherine.[11][12] SNAIL- en ZEB-factoren binden aan E-box-consensussequenties op het promotorgebied, terwijl KLF8 via GT-boxen aan de promotor bindt. Deze EMT-TF's onderdrukken niet alleen E-cadherine direct, maar onderdrukken ook transcriptioneel andere verbindingseiwitten, waaronder claudinen en desmosomen, waardoor EMT wordt vergemakkelijkt. Aan de andere kant worden transcriptiefactoren zoals grainyhead-achtige proteïne 2-homoloog (GRHL2) en ETS-gerelateerde transcriptiefactoren ELF3 en ELF5 gedownreguleerd tijdens EMT en blijken ze actief MET te stimuleren wanneer ze tot overexpressie worden gebracht in mesenchymcellen..[13][14] Sinds EMT bij de progressie van kanker EMT herovert in ontwikkelingsprogramma's, zijn veel van de EMT-TF's betrokken bij het bevorderen van metastatische gebeurtenissen.[15][16]
Verschillende signaalroutes (TGF-β, FGF, EGF, HGF, Wnt/bèta-catenine en Notch) en hypoxie kunnen EMT veroorzaken.[7][17][18] In het bijzonder is aangetoond dat Ras-MAPK Snail en Slug activeert.[19][20][21] Slug activeert de stappen van desmosomale loslaten, celverspreiding en gedeeltelijke scheiding aan de cel-celgrenzen, die de eerste en noodzakelijke fase van het EMT-proces vormen. Aan de andere kant kan Slug de tweede fase niet activeren,[22] die de inductie van celbeweeglijkheid, onderdrukking van de cytokeratine-expressie en activering van vimentine-expressie omvat.[23] Het is bekend dat Snail en Slug de expressie van p63-isovormen reguleren, een andere transcriptiefactor die nodig is voor een goede ontwikkeling van epitheelstructuren.[24] De veranderde expressie van p63-isovormen verminderde de cel-celadhesie en verhoogde de migratie-eigenschappen van kankercellen. De p63-factor is betrokken bij het remmen van EMT en de reductie van bepaalde p63-isovormen kan belangrijk zijn bij de ontwikkeling van epitheelkankers.[25] Van sommigen van hen is bekend dat ze de expressie van cytokeratines reguleren.[26] Er wordt ook gesuggereerd dat de fosfatidylinositol 3'-kinase (PI3K)/AKT-as, de Hedgehog-signaalroute, de celkernfactor NF-κB en de activerende transcriptiefactor 2 betrokken zijn bij EMT.[27][28][29][30]
Het Wnt-signaalreactiepad reguleert EMT bij gastrulatie, hartklepvorming en kanker.[31] Activering van het Wnt-reactiepad in borstkankercellen induceert de EMT-regulator Snail en upreguleert de mesenchymmerker vimentine. Ook correleert het actieve Wnt/bèta-catenine-reactiepad met een slechte prognose bij borstkankerpatiënten in de kliniek. Op dezelfde manier activeert TGF-β de expressie van Snail en ZEB om EMT bij de hartontwikkeling, palatogenese en kanker te reguleren. De botmetastase van borstkanker heeft de TGF-β-signalering geactiveerd, wat bijdraagt aan de vorming van deze laesies.[32] Aan de andere kant onderdrukt p53, een bekende tumorsuppressor, EMT door de expressie van verschillende microRNA's te activeren – miR-200 en miR-34 die de productie van de eiwitten ZEB en Snail remmen, en zo het epithele fenotype behouden.[33]
Ontwikkeling en wondgenezing
[bewerken | brontekst bewerken]Na de eerste fase van de embryogenese worden de innesteling van het embryo en de initiatie van de placentavorming geassocieerd met EMT. De trofoectoderm-cellen ondergaan EMT om de innesteling in het endometrium en de juiste plaatsing van de placenta te vergemakkelijken, waardoor de uitwisseling van voedingsstoffen en gasuitwisseling naar het embryo mogelijk wordt. Later in de embryogenese, tijdens de gastrulatie, zorgt EMT ervoor dat de cellen een specifiek gebied van het embryo kunnen binnendringen: de primitieve streep bij Amniota en de ventrale groef bij Drosophila. De cellen in dit weefsel brengen E-cadherine en apicaal-basale polariteit tot expressie.[34] Omdat gastrulatie een zeer snel proces is, wordt E-cadherine transcriptioneel onderdrukt door Twist en SNAI1 (gewoonlijk Snail genoemd), en op eiwitniveau door met P38 interacterend eiwit. De primitieve streep genereert, door invaginatie, verder mesoendoderm, dat zich scheidt om een mesoderm en een endoderm te vormen, opnieuw via EMT. Mesenchymale cellen uit de primitieve streep nemen ook deel aan de vorming van veel epitheel mesodermale organen, zoals de chorda dorsalis en somieten, via het omgekeerde van EMT, dwz mesenchymale-epitheliale overgang (MET). Lancetvisjes vormen een epitheliale neurale buis en een dorsaal notochord, maar hebben niet het EMT-potentieel van de primitieve streep. Bij hogere chordadieren komt het mesenchym voort uit de primitieve streep en migreert naar voren om de somieten te vormen en neemt samen met het mesenchym van de neurale top deel aan de vorming van het hartmesoderm.
Bij de gewervelden zijn epitheel en mesenchym de fundamentele weefselfenotypen. Tijdens de embryonale ontwikkeling worden migrerende neurale lijstcellen gegenereerd door EMT waarbij de epitheelcellen van het neuro-ectoderm betrokken zijn. Als gevolg hiervan scheiden deze cellen zich van de neurale plooien af, krijgen ze beweeglijkheid en verspreiden ze zich naar verschillende delen van het embryo, waar ze differentiëren naar vele andere celtypen. Ook wordt het craniofaciale mesenchym dat het bindweefsel vormt dat het hoofd en het gezicht vormt, door EMT uit neuralebuisepitheel gevormd.[35] EMT vindt plaats tijdens de vorming van de wervelkolom uit de extracellulaire matrix, die moet worden gesynthetiseerd door fibroblasten en osteoblasten die de neurale buis omringen. De belangrijkste bron van deze cellen zijn het sclerotoom en somietmesenchym, evenals de primitieve streep. Door de mesenchymale morfologie kunnen de cellen naar specifieke doelen in het embryo bewegen, waar ze differentiëren en/of differentiatie van andere cellen induceren.[35][36]
Tijdens wondgenezing ondergaan keratinocyten aan de rand van de wond EMT en ondergaan ze opnieuw epithelialisatie of MET wanneer de wond gesloten is. De expressie van Snail2 aan het migratiefront beïnvloedt deze toestand, omdat de overexpressie ervan de wondgenezing versnelt. Op dezelfde manier ondergaat het oppervlakte-epitheel van de eierstokken tijdens elke menstruatiecyclus EMT tijdens post-ovulatoire wondgenezing.[37]
Onvolledige EMT of een hybride E/M-fenotype
[bewerken | brontekst bewerken]Niet alle cellen ondergaan een volledige EMT, dat wil zeggen dat ze hun cel-celadhesie verliezen en solitaire migratie-eigenschappen verkrijgen. In plaats daarvan ondergaan de meeste cellen een onvolledige EMT, een toestand waarin ze enkele epitheel eigenschappen behouden, zoals cel-celadhesie of apico-basale polariteit, en migratie eigenschappen krijgen. Daarom hebben cellen in dit hybride epitheliale/mesenchymale (E/M) fenotype bijzondere eigenschappen zoals collectieve celmigratie.[38][39][40][31][41][42][43][44] Het volgen van eencellige cellen draagt bij aan het mogelijk maken van de visualisatie van morfologische overgangen tijdens EMT, het onderscheiden van fenotypes van celmigratie en de correlatie van de erfelijkheid van deze eigenschappen tussen zustercellen.[45] Er zijn twee wiskundige modellen voorgesteld, die proberen de opkomst van dit hybride E/M-fenotype te verklaren,[41][43] en het is zeer waarschijnlijk dat verschillende cellijnen verschillende hybride toestanden aannemen, zoals blijkt uit experimenten in MCF10A, HMLE en H1975-cellijnen.[42][46] Hoewel naar een hybride E/M-toestand wordt verwezen als 'metastabiel' of van voorbijgaande aard, suggereren recente experimenten in H1975-cellen dat deze toestand stabiel kan zijn.[47]
Kanker
[bewerken | brontekst bewerken]EMT bij kankerprogressie en metastase
[bewerken | brontekst bewerken]Het initiëren van metastasen vereist binnendringing, die mogelijk wordt gemaakt door EMT.[48][49] Carcinoomcellen in een primaire tumor verliezen cel-celadhesie gemedieerd door E-cadherine-repressie en breken door het basaal membraan met verhoogde binnendringingseigenschappen, en dringen de bloedbaan binnen. Later, wanneer deze circulerende tumorcellen (CTC's) de bloedbaan verlaten om micro-metastasen te vormen, ondergaan ze mesenchymale-epitheliale overgang (MET) voor klonale uitgroei op deze metastatische plaatsen. EMT en MET vormen dus de initiatie en voltooiing van de binnendringing-metastasecascade.[50] Op deze nieuwe metastase plaats kan de tumor andere processen ondergaan om de groei te optimaliseren. EMT is bijvoorbeeld in verband gebracht met PD-L1-expressie, vooral bij longkanker. Verhoogde niveaus van PD-L1 onderdrukken het immuunsysteem waardoor de kanker zich gemakkelijker kan verspreiden.[51]
EMT verleent resistentie tegen door oncogenen geïnduceerde voortijdige veroudering. Twist1 en Twist2, evenals ZEB1, beschermen menselijke cellen en embryonale fibroblasten van muizen tegen veroudering. Op vergelijkbare wijze kan TGF-β tumorbinnendringing en ontwijking van immuunbewaking in gevorderde stadia bevorderen. Wanneer TGF-β inwerkt op geactiveerde borstepitheelcellen die Ras tot expressie brengen, heeft EMT de voorkeur en wordt apoptose geremd.[52] Dit effect kan worden omgekeerd door inductoren van epitheeldifferentiatie, zoals GATA-3..[53]
Er is aangetoond dat EMT wordt geïnduceerd door androgeendeprivatietherapie bij metastatische prostaatkanker.[15] Activering van EMT-programma's via remming van de androgeen-as biedt een mechanisme waarmee tumorcellen zich kunnen aanpassen om herhaling en progressie van de ziekte te bevorderen. Brachyury, Axl, MEK en Aurora kinase A zijn moleculaire aanjagers van deze programma's, en remmers worden momenteel in klinische onderzoeken uitgevoerd om therapeutische toepassingen te bepalen.[15] Oncogene PKC-iota kan de binnendringing van melanoomcellen bevorderen door vimentine tijdens EMT te activeren. PKC-iota-remming of knockdown resulteerde in een toename van de E-cadherine- en RhoA-niveaus, terwijl de totale vimentine, gefosforyleerd vimentine (S39) en Par6 in metastatische melanoomcellen daalden. Deze resultaten suggereerden dat PKC-iota betrokken is bij signaalroutes die EMT bij melanoom upreguleren.[54][55]
Er zijn aanwijzingen dat EMT betrokken is bij het verwerven van resistentie tegen geneesmiddelen. Er werd gevonden dat de toename van EMT-merkers verband houdt met de resistentie van epitheelcellijnen van ovariumcarcinoom tegen paclitaxel. Op dezelfde manier verleent Snail ook resistentie tegen paclitaxel, adriamycine en bestraling door p53-gemedieerde apoptose te remmen.[56] Bovendien werd onlangs aangetoond dat ontstekingen, die in verband zijn gebracht met de progressie van kanker en fibrose, verband houden met kanker door middel van door ontstekingen geïnduceerde EMT.[57] Bijgevolg stelt EMT cellen in staat een migrerend fenotype te krijgen, evenals meervoudige immunosuppressie (remmen afweersysteem), resistentie tegen geneesmiddelen en het ontwijken van apoptose-mechanismen.
Er zijn aanwijzingen dat cellen die EMT ondergaan stamcelachtige eigenschappen krijgen, waardoor kankerstamcellen (CSC's) ontstaan. Na transfectie door geactiveerde Ras neemt een subpopulatie van cellen, die de vermeende stamcelmerkers CD44high/CD24low vertonen toe met de gelijktijdige inductie van EMT.[58] Ook is ZEB1 in staat stamcelachtige eigenschappen te verlenen, waardoor de relatie tussen EMT en stamcellen wordt versterkt. EMT kan dus een groter gevaar opleveren voor kankerpatiënten, omdat EMT er niet alleen voor zorgt dat de carcinoomcellen in de bloedbaan terechtkomen, maar ze ook eigenschappen van stamcellen geeft die het tumorverwekkende en proliferatie potentieel vergroten.[59]
Recente onderzoeken hebben de primaire effecten van EMT echter verder verschoven van binnendringing en metastase naar resistentie tegen chemotherapeutischee middelen. Onderzoek naar borstkanker en pancreaskanker toonde beide geen verschil aan in het metastatische potentieel van cellen bij verwerving van EMT.[60][61] Deze komen overeen met een andere studie die aantoont dat de EMT-transcriptiefactor TWIST feitelijk intacte zonula adhaerens vereist om lokale binnendringing bij borstkanker te bemiddelen.[38] De effecten van EMT en de relatie ervan met binnendringing en metastase kunnen daarom zeer contextspecifiek zijn.
In blaaskankercellijnen remt overexpressie van HDAC5 de proliferatie op de lange termijn, maar kan de epitheel-mesenchymale overgang (EMT) bevorderen.[62]
Bloedplaatjes-inductie van EMT in kankercellen
[bewerken | brontekst bewerken]
Bloedplaatjes hebben het vermogen om de inductie van EMT in kankercellen te initiëren. Wanneer bloedplaatjes naar een plaats in het bloedvat worden gebracht, kunnen ze een verscheidenheid aan groeifactoren vrijgeven (PDGF,[63] VEGF,[64] Angiopoietine-1[65]) en cytokinen, waaronder de EMT-inductor TGF-β.[66] De afgifte van TGF-β door bloedplaatjes in bloedvaten nabij primaire tumoren verhoogt de indringing en bevordert de metastase van kankercellen in de tumor.[67] Studies naar defecte bloedplaatjes en een verminderd aantal bloedplaatjes in muismodellen hebben aangetoond dat een verminderde bloedplaatjesfunctie samengaat met verminderde metastatische vorming.[68][69] Bij mensen zijn het aantal bloedplaatjes en trombocytose (een verhoogd aantal bloedplaatjes in het bloed) in verband gebracht met gevorderde, vaak metastatische kanker bij baarmoederhalskanker,[70] eierstokkanker,[71] maagkanker[72] en slokdarmkanker.[73] Hoewel er veel onderzoek is gedaan naar het bestuderen van interacties tussen tumorcellen en bloedplaatjes, is er nog geen kankertherapie ontwikkeld die zich op deze interactie richt.[74] Dit kan gedeeltelijk te wijten zijn aan de herhaling van protrombotische reactiepaden die het gebruik van meerdere therapeutische benaderingen zouden vereisen om pro-metastatische gebeurtenissen te voorkomen via EMT-inductie in kankercellen door geactiveerde bloedplaatjes.
Om de kansen op de ontwikkeling van een kankermetastase te vergroten, moet een kankercel detectie en waarneming door het immuunsysteem vermijden zodra deze in de bloedbaan terechtkomt. Geactiveerde bloedplaatjes hebben het vermogen om glycoproteïnen en glycolipiden (P-selectineliganden zoals PSGL-1) op het oppervlak van kankercellen te binden voor een fysieke barrière die de kankercel beschermt tegen door natuurlijke killercellen gemedieerde lyse in de bloedbaan.[75] Bovendien bevorderen geactiveerde bloedplaatjes de adhesie van kankercellen aan geactiveerde endotheelcellen die de bloedvaten bekleden met behulp van adhesiemoleculen die op bloedplaatjes aanwezig zijn.[76][74] P-selectine liganden op het oppervlak van kankercellen moeten nog worden opgehelderd en kunnen dienen als potentiële biomarkers voor ziekteprogressie bij kanker.[74]
Geneesmiddelen gericht op EMT tegen kanker
[bewerken | brontekst bewerken]Veel studies hebben voorgesteld dat inductie van EMT het primaire mechanisme is waardoor epitheelkankercellen kwaadaardige fenotypes worden die metastase bevorderen.[77] Geneesmiddelenontwikkeling gericht op de activering van EMT in kankercellen is dus een doel geworden van farmaceutische bedrijven.[78]
Micromoleculen die door TGF-β geïnduceerde EMT kunnen remmen, zijn in ontwikkeling.[78] Silmitasertib (CX-4945) is een micromolecuulremmer van proteïnekinase CK2, die in verband wordt gebracht met door TGF-β geïnduceerde EMT, en bevindt zich momenteel in klinische onderzoeken voor cholangiocarcinoom (galwegkanker), evenals in preklinische ontwikkeling voor hematologische en lymfoïde malignes.[79][80] In januari 2017 kreeg Silmitasertib de status van weesgeneesmiddel van de Amerikaanse Food and Drug Administration voor cholangiocarcinoom en bevindt zich momenteel in een fase II-studie. Klinische onderzoeken om vermeende voordelen aan te tonen zijn nog aan de gang.[81] Silmitasertib wordt ontwikkeld door Senhwa Biosciences.[82] Een andere micromoleculaire remmer Galunisertib (LY2157299) is een krachtige TGF-β type I-receptorkinaseremmer waarvan is aangetoond dat deze de grootte, de groeisnelheid van tumoren en het tumorvormende potentieel vermindert in drievoudige negatieve borstkankercellijnen met behulp van muizenxenotransplantaten.[83] Galunisertib werd ontwikkeld door Lilly Oncology en bevond zich in fase I/II klinische onderzoeken voor hepatocellulair carcinoom, niet door een chirurgische ingreep verwijderbare pancreaskanker en kwaadaardig glioom.[84] In januari 2020 is Eli Lilly met het onderzoek gestopt.[85] Er wordt gesuggereerd dat mnicromolecuulremmers van EMT niet fungeren als vervanging voor traditionele chemotherapeutische middelen, maar waarschijnlijk de grootste werkzaamheid zullen vertonen bij de behandeling van kanker wanneer ze in combinatie daarmee worden gebruikt.
Antagomirs en microRNA-nabootsers hebben belangstelling gekregen als potentiële bron van therapieën om EMT-geïnduceerde metastase bij kanker aan te pakken en om vele andere ziekten te behandelen.[86] Antagomirs, ook bekend als anti-miRs, zijn een klasse van chemisch gemanipuleerde oligonucleotiden die zijn ontworpen om microRNA's tot zwijgen te brengen. Antagomirs werden voor het eerst ontwikkeld om zich te richten op miR-122, een microRNA dat overvloedig aanwezig was en specifiek was voor de lever. Deze ontdekking heeft geleid tot de ontwikkeling van andere antagomirs die kunnen koppelen aan specifieke microRNA's die aanwezig zijn in de micro-omgeving van de tumor of in de kankercellen.[87][84] Een microRNA dat miR-655 nabootst, bleek EMT te onderdrukken door het richten van EMT-inducerende transcriptiefactor ZEB1 en TGF-β-receptor 2 in een pancreaskankercellijn. Overexpressie van de miR-655-nabootser in de Panc1-kankercellijn verhoogde de expressie van E-cadherine en onderdrukte de migratie en indringing van mesenchymachtige kankercellen.[88] Het gebruik van microRNA-nabootsers om EMT te onderdrukken is uitgebreid naar andere kankercellijnen en biedt potentieel voor de ontwikkeling van klinische geneesmiddelen.[86] MicroRNA-nabootsers en antagomirs lijden echter aan een gebrek aan stabiliteit in vivo en missen een nauwkeurig afgiftesysteem om deze moleculen voor behandeling naar de tumorcellen of het weefsel te sturen.[89] Verbeteringen aan de antagomir- en microRNA-nabootsende stabiliteit door chemische modificaties zoals vergrendelde nucleïnezuur (LNA) oligonucleotiden of peptidenucleïnezuren (PNA) kunnen de snelle afbraak van deze kleine moleculen door RNasen voorkomen.[89][86] De levering van antagomirs en microRNA-nabootsers in cellen door deze moleculen in liposoom-nanodeeltjes in te sluiten heeft interesse gewekt, maar liposoomstructuren lijden aan hun eigen nadelen die moeten worden overwonnen voor hun effectieve gebruik als medicijnafgiftemechanisme.[89] Deze nadelen van liposoom-nanodeeltjes omvatten niet-specifieke opname door cellen en inductie van immuunreacties.[90] De rol die microRNA's spelen bij de ontwikkeling en metastase van kanker wordt wetenschappelijk veel onderzocht en het moet nog worden aangetoond of microRNA-nabootsers of antagomirs kunnen dienen als standaard klinische behandelingen om EMT of oncogene microRNA's bij kanker te onderdrukken.[86]
Generatie van endocriene voorlopercellen uit eilandjes van Langerhans
[bewerken | brontekst bewerken]Vergelijkbaar met het genereren van kankerstamcellen, werd aangetoond dat EMT-voorlopercellen genereert konden worden uit menselijke eilandjes van Langerhans.[91] Aanvankelijk werd voorgesteld dat de van menselijke eilandjes van Langerhans afkomstige voorlopercellen (hIPC's) betere voorlopers zijn, aangezien β-cel-nakomelingen in deze hIPC's epigenetische kenmerken erven die een actief insulinepromotergebied definiëren.[92] Later suggereerde een andere reeks experimenten echter dat gelabelde β-cellen in vitro dedifferentiëren tot een mesenchymaalachtig fenotype, maar niet prolifereren; waarmee in 2007 een debat op gang werd gebracht.[93][94][95]
Omdat deze onderzoeken bij menselijke eilandjes van Langerhans geen analyse van de afstamming bevatten, werden deze bevindingen van onomkeerbaar gelabelde β-cellen bij muizen geëxtrapoleerd naar menselijke eilandjes van Langerhans. Door gebruik te maken van een tweetallig lentiviraal en genetisch afstammingssysteem om β-cellen te labelen, werd op overtuigende wijze aangetoond dat β-cellen van volwassen menselijke eilandjes van Langerhans EMT ondergaan en in vitro prolifereren..[96][97] Deze bevindingen werden ook bevestigd in menselijke foetale insulineproducerende cellen van de alvleesklier, en de mesenchymale cellen afgeleid van de eilandjes van Langerhans kunnen het omgekeerde van EMT ondergaan – MET – om eilandjesachtige celaggregaten te genereren.[98] Het concept van het genereren van voorlopercellen uit insulineproducerende cellen door EMT of het genereren van kankerstamcellen tijdens EMT bij kanker kan dus potentie hebben voor vervangingstherapie bij diabetes en pleit voor medicijnen die zich richten op de remming van EMT bij kanker.[98]
Afbeeldingen
[bewerken | brontekst bewerken]-
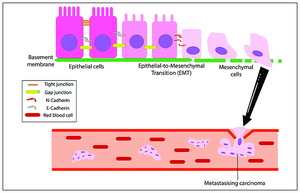 Het EMT-proces naar kanker
Het EMT-proces naar kanker -
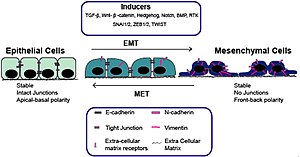 Epitheel-mesenchymale overgang (EMT) met inductoren
Epitheel-mesenchymale overgang (EMT) met inductoren -
 Verschillende graden van epitheel-mesenchymale overgang (EMT) correleren met verschillende weefselmorfologieën
Verschillende graden van epitheel-mesenchymale overgang (EMT) correleren met verschillende weefselmorfologieën -
 Ingressie bij een vogel-embryo
Ingressie bij een vogel-embryo




- ↑ Kong D, Li Y, Wang Z, Sarkar FH (February 2011). Cancer Stem Cells and Epithelial-to-Mesenchymal Transition (EMT)-Phenotypic Cells: Are They Cousins or Twins?. Cancers 3 (1): 716–29. PMID 21643534. PMC 3106306. DOI: 10.3390/cancers30100716.
- ↑ Lamouille S, Xu J, Derynck R (March 2014). Molecular mechanisms of epithelial-mesenchymal transition. Nature Reviews. Molecular Cell Biology 15 (3): 178–96. PMID 24556840. PMC 4240281. DOI: 10.1038/nrm3758.
- ↑ Thiery JP, Acloque H, Huang RY, Nieto MA (November 2009). Epithelial-meedsenchymal transitions in development and disease. Cell 139 (5): 871–90. PMID 19945376. DOI: 10.1016/j.cell.2009.11.007.
- ↑ Thiery JP, Sleeman JP (February 2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nature Reviews. Molecular Cell Biology 7 (2): 131–42. PMID 16493418. DOI: 10.1038/nrm1835.
- ↑ Francou A, Anderson KV (2020). The Epithelial-to-Mesenchymal Transition in Development and Cancer. Annual Review of Cancer Biology 4: 197–220. PMID 34113749. PMC 8189433. DOI: 10.1146/annurev-cancerbio-030518-055425.
- ↑ Phua YL, Martel N, Pennisi DJ, Little MH, Wilkinson L (April 2013). Distinct sites of renal fibrosis in Crim1 mutant mice arise from multiple cellular origins. The Journal of Pathology 229 (5): 685–96. PMID 23224993. DOI: 10.1002/path.4155.
- ↑ a b Kalluri R, Weinberg RA (June 2009). The basics of epithelial-mesenchymal transition. The Journal of Clinical Investigation 119 (6): 1420–8. PMID 19487818. PMC 2689101. DOI: 10.1172/JCI39104.
- ↑ Sciacovelli M, Frezza C (October 2017). Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. The FEBS Journal 284 (19): 3132–3144. PMID 28444969. PMC 6049610. DOI: 10.1111/febs.14090.
- ↑ Li L, Li W (June 2015). Epithelial-mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacology & Therapeutics 150: 33–46. PMID 25595324. DOI: 10.1016/j.pharmthera.2015.01.004.
- ↑ a b c Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587.
- ↑ Peinado H, Olmeda D, Cano A (2007). Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype?. Nature Reviews Cancer 7 (6): 415–428. PMID 17508028. DOI: 10.1038/nrc2131.
- ↑ Yang J, Weinberg RA (2008). Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14 (6): 818–829. PMID 18539112. DOI: 10.1016/j.devcel.2008.05.009.
- ↑ De Craene B, Berx G (2013). Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews Cancer 13 (2): 97–110. PMID 23344542. DOI: 10.1038/nrc3447.
- ↑ Chakrabarti R, Hwang J, Andres Blanco M, Wei Y, Lukačišin M, Romano RA, Smalley K, Liu S, Yang Q, Ibrahim T, Mercatali L, Amadori D, Haffty BG, Sinha S, Kang Y (2012). Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat Cell Biol 14 (11): 1212–1222. PMID 23086238. PMC 3500637. DOI: 10.1038/ncb2607.
- ↑ a b c Nouri M, Ratther E, Stylianou N, Nelson CC, Hollier BG, Williams ED (2014). Androgen-targeted therapy-induced epithelial mesenchymal plasticity and neuroendocrine transdifferentiation in prostate cancer: an opportunity for intervention. Front Oncol 4: 370. PMID 25566507. PMC 4274903. DOI: 10.3389/fonc.2014.00370.
- ↑ Puisieux A, Brabletz T, Caramel J (June 2014). Oncogenic roles of EMT-inducing transcription factors. Nature Cell Biology 16 (6): 488–94. PMID 24875735. DOI: 10.1038/ncb2976.
- ↑ Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, Liu J, Wang Q, Zhu J, Feng X, Dong J, Qian C (March 2013). Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor -1α in hepatocellular carcinoma. BMC Cancer 13: 108. PMID 23496980. PMC 3614870. DOI: 10.1186/1471-2407-13-108.
- ↑ Epithelial-Mesenchymal Transition | GeneTex. www.genetex.com. Geraadpleegd op 28 oktober 2019.
- ↑ Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M (January 2009). Role of Ras signaling in the induction of snail by transforming growth factor-beta. The Journal of Biological Chemistry 284 (1): 245–53. PMID 19010789. DOI: 10.1074/jbc.m804777200.
- ↑ Ciruna B, Rossant J (July 2001). FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Developmental Cell 1 (1): 37–49. PMID 11703922. DOI: 10.1016/s1534-5807(01)00017-x.
- ↑ Lu Z, Ghosh S, Wang Z, Hunter T (December 2003). Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell 4 (6): 499–515. PMID 14706341. DOI: 10.1016/s1535-6108(03)00304-0.
- ↑ Savagner P, Yamada KM, Thiery JP (June 1997). The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. The Journal of Cell Biology 137 (6): 1403–19. PMID 9182671. PMC 2132541. DOI: 10.1083/jcb.137.6.1403.
- ↑ Boyer B, Tucker GC, Vallés AM, Franke WW, Thiery JP (October 1989). Rearrangements of desmosomal and cytoskeletal proteins during the transition from epithelial to fibroblastoid organization in cultured rat bladder carcinoma cells. The Journal of Cell Biology 109 (4 Pt 1): 1495–509. PMID 2677020. PMC 2115780. DOI: 10.1083/jcb.109.4.1495.
- ↑ Herfs M, Hubert P, Suarez-Carmona M, Reschner A, Saussez S, Berx G, Savagner P, Boniver J, Delvenne P (April 2010). Regulation of p63 isoforms by snail and slug transcription factors in human squamous cell carcinoma. The American Journal of Pathology 176 (4): 1941–9. PMID 20150431. PMC 2843482. DOI: 10.2353/ajpath.2010.090804.
- ↑ Lindsay J, McDade SS, Pickard A, McCloskey KD, McCance DJ (February 2011). Role of DeltaNp63gamma in epithelial to mesenchymal transition. The Journal of Biological Chemistry 286 (5): 3915–24. PMID 21127042. PMC 3030392. DOI: 10.1074/jbc.M110.162511.
- ↑ Boldrup L, Coates PJ, Gu X, Nylander K (December 2007). DeltaNp63 isoforms regulate CD44 and keratins 4, 6, 14 and 19 in squamous cell carcinoma of head and neck. The Journal of Pathology 213 (4): 384–91. PMID 17935121. DOI: 10.1002/path.2237.
- ↑ Larue L, Bellacosa A (November 2005). Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene 24 (50): 7443–54. PMID 16288291. DOI: 10.1038/sj.onc.1209091.
- ↑ Vlahopoulos SA, Logotheti S, Mikas D, Giarika A, Gorgoulis V, Zoumpourlis V (April 2008). The role of ATF-2 in oncogenesis. BioEssays 30 (4): 314–27. PMID 18348191. DOI: 10.1002/bies.20734.
- ↑ Huber MA, Beug H, Wirth T (December 2004). Epithelial-mesenchymal transition: NF-kappaB takes center stage. Cell Cycle 3 (12): 1477–80. PMID 15539952. DOI: 10.4161/cc.3.12.1280.
- ↑ Katoh Y, Katoh M (September 2008). Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review). International Journal of Molecular Medicine 22 (3): 271–5. PMID 18698484.
- ↑ a b Micalizzi DS, Farabaugh SM, Ford HL (2010). Epithelial-Mesenchymal Transition in Cancer: Parallels between Normal Development and Tumor Progression. J Mammary Gland Biol Neoplasia 15 (2): 117–134. PMID 20490631. PMC 2886089. DOI: 10.1007/s10911-010-9178-9.
- ↑ Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, Manova-Todorova K, Blasberg R, Gerald WL, Massagué J (2005). Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. PNAS 102 (39): 13909–14. PMID 16172383. PMC 1236573. DOI: 10.1073/pnas.0506517102.
- ↑ Chang C, Chao C, Xia W, Yang J, Xiong Y, Li C, Yu W, Rehman SK, Hsu JL, Lee H, Liu M, Chen C, Yu D, Hung M (2011). p53 regulates epithelial-mesenchymal transition (EMT) and stem cell properties through modulating miRNAs. Nat Cell Biol 13 (3): 317–323. PMID 21336307. PMC 3075845. DOI: 10.1038/ncb2173.
- ↑ Lim R, Thiery JP (2012). Epithelial-mesenchymal transitions: insights from development. Development 139 (19): 3471–3486. PMID 22949611. DOI: 10.1242/dev.071209.
- ↑ a b Hay ED (2005). The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev. Dyn. 233 (3): 706–20. PMID 15937929. DOI: 10.1002/dvdy.20345.
- ↑ Kerosuo L, Bronner-Fraser M (2012). What is bad in cancer is good in the embryo: Importance of EMT in neural crest development. Seminars in Cell and Developmental Biology 23 (3): 320–332. PMID 22430756. PMC 3345076. DOI: 10.1016/j.semcdb.2012.03.010.
- ↑ Ahmed N, Maines-Bandiera S, Quinn MA, Unger WG, Dedhar S, Auersperg N (2006). Molecular pathways regulating EGF-induced epithelio- mesenchymal transition in human ovarian surface epithelium. Am J Physiol Cell Physiol 290 (6): C1532–C1542. PMID 16394028. DOI: 10.1152/ajpcell.00478.2005.
- ↑ a b Shamir ER, Pappalardo E, Jorgens DM, Coutinho K, Tsai WT, Aziz K, Auer M, Tran PT, Bader JS, Ewald AJ (March 2014). Twist1-induced dissemination preserves epithelial identity and requires E-cadherin. The Journal of Cell Biology 204 (5): 839–56. PMID 24590176. PMC 3941052. DOI: 10.1083/jcb.201306088.
- ↑ Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben-Jacob E, Onuchic JN, Levine H (1 januari 2015). Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Frontiers in Oncology 5: 155. PMID 26258068. PMC 4507461. DOI: 10.3389/fonc.2015.00155.
- ↑ Nakaya Y, Sheng G (November 2013). EMT in developmental morphogenesis. Cancer Letters 341 (1): 9–15. PMID 23462225. DOI: 10.1016/j.canlet.2013.02.037.
- ↑ a b Tian XJ, Zhang H, Xing J (August 2013). Coupled reversible and irreversible bistable switches underlying TGFβ-induced epithelial to mesenchymal transition. Biophysical Journal 105 (4): 1079–89. PMID 23972859. PMC 3752104. DOI: 10.1016/j.bpj.2013.07.011.
- ↑ a b Zhang J, Tian XJ, Zhang H, Teng Y, Li R, Bai F, Elankumaran S, Xing J (September 2014). TGF-β-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Science Signaling 7 (345): ra91. PMID 25270257. DOI: 10.1126/scisignal.2005304.
- ↑ a b Lu M, Jolly MK, Levine H, Onuchic JN, Ben-Jacob E (November 2013). MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proceedings of the National Academy of Sciences of the United States of America 110 (45): 18144–9. PMID 24154725. PMC 3831488. DOI: 10.1073/pnas.1318192110.
- ↑ Savagner P (October 2010). The epithelial-mesenchymal transition (EMT) phenomenon. Annals of Oncology 21 (Suppl 7): vii89-92. PMID 20943648. PMC 3379967. DOI: 10.1093/annonc/mdq292.
- ↑ (en) Quinsgaard, Ellen Marie Botne, Korsnes, Mónica Suárez, Korsnes, Reinert, Moestue, Siver Andreas (April 2024). Single-cell tracking as a tool for studying EMT-phenotypes. Experimental Cell Research 437 (1): 113993. DOI: 10.1016/j.yexcr.2024.113993.
- ↑ Jia D, Jolly MK, Tripathi SC, Den Hollander P, Huang B, Lu M, Celiktas M, Ramirez-Peña E, Ben-Jacob E, Onuchic JN, Hanash SM, Mani SA, Levine H (2017). Distinguishing mechanisms underlying EMT tristability. Cancer Convergence 1 (1): 2. PMID 29623961. PMC 5876698. DOI: 10.1186/s41236-017-0005-8.
- ↑ Jolly MK, Tripathi SC, Jia D, Mooney SM, Celiktas M, Hanash SM, Mani SA, Pienta KJ, Ben-Jacob E, Levine H (May 2016). Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 7 (19): 27067–84. PMID 27008704. PMC 5053633. DOI: 10.18632/oncotarget.8166.
- ↑ Hanahan D, Weinberg RA (January 2000). The hallmarks of cancer. Cell 100 (1): 57–70. PMID 10647931. DOI: 10.1016/s0092-8674(00)81683-9.
- ↑ Hanahan D, Weinberg RA (March 2011). Hallmarks of cancer: the next generation. Cell 144 (5): 646–74. PMID 21376230. DOI: 10.1016/j.cell.2011.02.013.
- ↑ Chaffer CL, Weinberg RA (March 2011). A perspective on cancer cell metastasis. Science 331 (6024): 1559–64. PMID 21436443. DOI: 10.1126/science.1203543.
- ↑ Ye X, Weinberg RA (November 2015). Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends in Cell Biology 25 (11): 675–686. PMID 26437589. PMC 4628843. DOI: 10.1016/j.tcb.2015.07.012.
- ↑ Massague J (2008). TGFβ in cancer. Cell 134 (2): 215–229. PMID 18662538. PMC 3512574. DOI: 10.1016/j.cell.2008.07.001.
- ↑ Chu IM, Lai WC, Aprelikova O, El Touny LH, Kouros-Mehr H, Green JE (2013). Expression of GATA3 in MDA-MB-231 triple-negative breast cancer cells induces a growth inhibitory response to TGFß. PLOS ONE 8 (4): e61125. PMID 23577196. PMC 3620110. DOI: 10.1371/journal.pone.0061125.
- ↑ Ratnayake WS, Apostolatos AH, Ostrov DA, Acevedo-Duncan M (2017). Two novel atypical PKC inhibitors; ACPD and DNDA effectively mitigate cell proliferation and epithelial to mesenchymal transition of metastatic melanoma while inducing apoptosis. Int. J. Oncol. 51 (5): 1370–1382. PMID 29048609. PMC 5642393. DOI: 10.3892/ijo.2017.4131.
- ↑ Ratnayake WS, Apostolatos CA, Apostolatos AH, Schutte RJ, Huynh MA, Ostrov DA, Acevedo-Duncan M (2018). Oncogenic PKC-ι activates Vimentin during epithelial-mesenchymal transition in melanoma; a study based on PKC-ι and PKC-ζ specific inhibitors. Cell Adhes. Migr. 12 (5): 447–463. PMID 29781749. PMC 6363030. DOI: 10.1080/19336918.2018.1471323.
- ↑ Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A, Kikkawa F (August 2007). Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. International Journal of Oncology 31 (2): 277–83. PMID 17611683. DOI: 10.3892/ijo.31.2.277.
- ↑ Ricciardi M, Zanotto M, Malpeli G, Bassi G, Perbellini O, Chilosi M, Bifari F, Krampera M (March 2015). Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. British Journal of Cancer 112 (6): 1067–75. PMID 25668006. PMC 4366889. DOI: 10.1038/bjc.2015.29.
- ↑ Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133 (4): 704–15. PMID 18485877. PMC 2728032. DOI: 10.1016/j.cell.2008.03.027.
- ↑ Singh A, Settleman J (2010). EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29 (34): 4741–4751. PMID 20531305. PMC 3176718. DOI: 10.1038/onc.2010.215.
- ↑ Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, Schwabe RF, Vahdat LT, Altorki NK, Mittal V, Gao D (November 2015). Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527 (7579): 472–6. PMID 26560033. PMC 4662610. DOI: 10.1038/nature15748.
- ↑ Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R (November 2015). Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527 (7579): 525–530. PMID 26560028. PMC 4849281. DOI: 10.1038/nature16064.
- ↑ Jaguva Vasudevan AA, Hoffmann MJ, Beck ML, Poschmann G, Petzsch P, Wiek C, Stühler K, Köhrer K, Schulz WA, Niegisch G (April 2019). HDAC5 Expression in Urothelial Carcinoma Cell Lines Inhibits Long-Term Proliferation but Can Promote Epithelial-to-Mesenchymal Transition. International Journal of Molecular Sciences 20 (9): 2135. PMID 31052182. PMC 6539474. DOI: 10.3390/ijms20092135.
- ↑ Kepner N, Lipton A (February 1981). A mitogenic factor for transformed fibroblasts from human platelets. Cancer Research 41 (2): 430–2. PMID 6256066.
- ↑ Möhle R, Green D, Moore MA, Nachman RL, Rafii S (January 1997). Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proceedings of the National Academy of Sciences of the United States of America 94 (2): 663–8. PMID 9012841. PMC 19570. DOI: 10.1073/pnas.94.2.663.
- ↑ Li JJ, Huang YQ, Basch R, Karpatkin S (February 2001). Thrombin induces the release of angiopoietin-1 from platelets. Thrombosis and Haemostasis 85 (2): 204–6. PMID 11246533. DOI: 10.1055/s-0037-1615677.
- ↑ Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB (June 1983). Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. The Journal of Biological Chemistry 258 (11): 7155–60. PMID 6602130. DOI: 10.1016/S0021-9258(18)32345-7.
- ↑ Oft M, Heider KH, Beug H (November 1998). TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Current Biology 8 (23): 1243–52. PMID 9822576. DOI: 10.1016/s0960-9822(07)00533-7.
- ↑ Bakewell SJ, Nestor P, Prasad S, Tomasson MH, Dowland N, Mehrotra M, Scarborough R, Kanter J, Abe K, Phillips D, Weilbaecher KN (November 2003). Platelet and osteoclast beta3 integrins are critical for bone metastasis. Proceedings of the National Academy of Sciences of the United States of America 100 (24): 14205–10. PMID 14612570. PMC 283570. DOI: 10.1073/pnas.2234372100.
- ↑ Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR (July 2004). Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 104 (2): 397–401. PMID 15031212. DOI: 10.1182/blood-2004-02-0434.
- ↑ Hernandez E, Lavine M, Dunton CJ, Gracely E, Parker J (June 1992). Poor prognosis associated with thrombocytosis in patients with cervical cancer. Cancer 69 (12): 2975–7. PMID 1591690. DOI: <2975::aid-cncr2820691218>3.0.co;2-a 10.1002/1097-0142(19920615)69:12<2975::aid-cncr2820691218>3.0.co;2-a.
- ↑ Zeimet AG, Marth C, Müller-Holzner E, Daxenbichler G, Dapunt O (February 1994). Significance of thrombocytosis in patients with epithelial ovarian cancer. American Journal of Obstetrics and Gynecology 170 (2): 549–54. PMID 8116711. DOI: 10.1016/s0002-9378(94)70225-x.
- ↑ Ikeda M, Furukawa H, Imamura H, Shimizu J, Ishida H, Masutani S, Tatsuta M, Satomi T (April 2002). Poor prognosis associated with thrombocytosis in patients with gastric cancer. Annals of Surgical Oncology 9 (3): 287–91. PMID 11923136. DOI: 10.1245/aso.2002.9.3.287.
- ↑ Shimada H, Oohira G, Okazumi S, Matsubara H, Nabeya Y, Hayashi H, Takeda A, Gunji Y, Ochiai T (May 2004). Thrombocytosis associated with poor prognosis in patients with esophageal carcinoma. Journal of the American College of Surgeons 198 (5): 737–41. PMID 15110807. DOI: 10.1016/j.jamcollsurg.2004.01.022.
- ↑ a b c Erpenbeck L, Schön MP (April 2010). Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood 115 (17): 3427–36. PMID 20194899. PMC 2867258. DOI: 10.1182/blood-2009-10-247296.
- ↑ Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Jirousková M, Degen JL (January 2005). Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 105 (1): 178–85. PMID 15367435. DOI: 10.1182/blood-2004-06-2272.
- ↑ Gay LJ, Felding-Habermann B (February 2011). Contribution of platelets to tumour metastasis. Nature Reviews. Cancer 11 (2): 123–34. PMID 21258396. PMC 6894505. DOI: 10.1038/nrc3004.
- ↑ Thiery JP (June 2002). Epithelial-mesenchymal transitions in tumour progression. Nature Reviews. Cancer 2 (6): 442–54. PMID 12189386. DOI: 10.1038/nrc822.
- ↑ a b Yingling JM, Blanchard KL, Sawyer JS (December 2004). Development of TGF-beta signalling inhibitors for cancer therapy. Nature Reviews. Drug Discovery 3 (12): 1011–22. PMID 15573100. DOI: 10.1038/nrd1580.
- ↑ Zou J, Luo H, Zeng Q, Dong Z, Wu D, Liu L (June 2011). Protein kinase CK2α is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes. Journal of Translational Medicine 9: 97. PMID 21702981. PMC 3132712. DOI: 10.1186/1479-5876-9-97.
- ↑ Gowda C, Sachdev M, Muthusami S, Kapadia M, Petrovic-Dovat L, Hartman M, Ding Y, Song C, Payne JL, Tan BH, Dovat S (2017). Casein Kinase II (CK2) as a Therapeutic Target for Hematological Malignancies. Current Pharmaceutical Design 23 (1): 95–107. PMID 27719640. DOI: 10.2174/1381612822666161006154311.
- ↑ Marilyn Glassberg, MD Silmitasertib (CX-4945) in Patients With Severe Coronavirus Disease 2019 (COVID-19) (CX4945). The University of Arizona
- ↑ (en) (10 maart 2017). CX-4945 Granted Orphan Drug Designation. Oncology Times 39 (5). ISSN: 0276-2234. DOI: 10.1097/01.cot.0000514203.35081.69.
- ↑ Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, Sanders M, Stanford J, Cook RS, Arteaga CL (March 2013). TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. The Journal of Clinical Investigation 123 (3): 1348–58. PMID 23391723. PMC 3582135. DOI: 10.1172/JCI65416.
- ↑ a b Kothari AN, Mi Z, Zapf M, Kuo PC (15 oktober 2014). Novel clinical therapeutics targeting the epithelial to mesenchymal transition. Clinical and Translational Medicine 3: 35. PMID 25343018. PMC 4198571. DOI: 10.1186/s40169-014-0035-0.
- ↑ Eli Lilly cuts 3 cancer drugs amid Q4 clear-out.
- ↑ a b c d Rupaimoole R, Slack FJ (March 2017). MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nature Reviews. Drug Discovery 16 (3): 203–222. PMID 28209991. DOI: 10.1038/nrd.2016.246.
- ↑ Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M (December 2005). Silencing of microRNAs in vivo with 'antagomirs'. Nature 438 (7068): 685–9. PMID 16258535. DOI: 10.1038/nature04303.
- ↑ Harazono Y, Muramatsu T, Endo H, Uzawa N, Kawano T, Harada K, Inazawa J, Kozaki K (14 mei 2013). miR-655 Is an EMT-suppressive microRNA targeting ZEB1 and TGFBR2. PLOS ONE 8 (5): e62757. PMID 23690952. PMC 3653886. DOI: 10.1371/journal.pone.0062757.
- ↑ a b c Rothschild SI (4 maart 2014). microRNA therapies in cancer. Molecular and Cellular Therapies 2: 7. PMID 26056576. PMC 4452061. DOI: 10.1186/2052-8426-2-7.
- ↑ Lv H, Zhang S, Wang B, Cui S, Yan J (August 2006). Toxicity of cationic lipids and cationic polymers in gene delivery. Journal of Controlled Release 114 (1): 100–9. PMID 16831482. DOI: 10.1016/j.jconrel.2006.04.014.
- ↑ Gershengorn MC, Hardikar AA, Wei C (2004). Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells. Science 306 (5705): 2261–2264. PMID 15564314. DOI: 10.1126/science.1101968.
- ↑ Gershengorn MC, Geras-Raaka E, Hardikar AA (2005). Are better islet cell precursors generated by epithelial-to-mesenchymal transition?. Cell Cycle 4 (3): 380–382. PMID 15711124. DOI: 10.4161/cc.4.3.1538.
- ↑ Atouf F, Park CH, Pechhold K (2007). No evidence for mouse pancreatic beta-cell epithelial-mesenchymal transition in vitro. Diabetes 56 (3): 699–702. PMID 17327438. DOI: 10.2337/db06-1446.
- ↑ Chase LG, Ulloa-Montoya F, Kidder BL (2007). Islet-derived fibroblast-like cells are not derived via epithelial-mesenchymal transition from Pdx-1 or insulin-positive cells. Diabetes 56 (1): 3–7. PMID 17110468. DOI: 10.2337/db06-1165.
- ↑ Morton RA, Geras-Raaka E, Wilson LM (2007). Endocrine precursor cells from mouse islets are not generated by epithelial-to-mesenchymal transition of mature beta cells. Mol Cell Endocrinol 270 (1–2): 87–93. PMID 17363142. PMC 1987709. DOI: 10.1016/j.mce.2007.02.005.
- ↑ Russ HA, Bar Y, Ravassard P (2008). In vitro proliferation of cells derived from adult human beta-cells revealed by cell-lineage tracing. Diabetes 57 (6): 1575–1583. PMID 18316362. DOI: 10.2337/db07-1283.
- ↑ Russ HA, Ravassard P, Kerr-Conte J (2009). Epithelial-mesenchymal transition in cells expanded in vitro from lineage-traced adult human pancreatic beta cells. PLOS ONE 4 (7). PMID 19641613. PMC 2712769. DOI: 10.1371/journal.pone.0006417.
- ↑ a b Joglekar MV, Joglekar VM, Joglekar SV (2009). Human fetal pancreatic insulin-producing cells proliferate in vitro. J Endocrinol 201 (1): 27–36. PMID 19171567. DOI: 10.1677/joe-08-0497.